


二代16S,长期以来一直是微生物群落分析的首选方法(Woese & Fox, 1977),然而,由于其PE150的测序读长,只能测16S rRNA基因的部分区域,如V3-V4区域,这对于某些具有高序列相似性的微生物群体,不能提供足够的分辨率来准确区分近缘物种(Caporaso et al., 2012)。
宏基因组测序提供了一种相对详实分析环境样品中所有基因的方法。尽管这种方法能够提供关于功能多样性的丰富信息,但其在物种水平的分辨率仍然受测序长度限制,且数据分析复杂,计算成本较高(Quince et al., 2017)。
三代16S全长测序技术能够揭示更为丰富的微生物群落结构,能够更有效地检测到低丰度的物种(Singer et al., 2016)。并且由于全长测序可提升物种分辨率,有助于揭示微生物群落结构的细微差别(Johnson et al., 2019)。
之前由于HiFi数据利用率的问题,对三代16S进行高深度的测序成本非常高,所以大部分的情况下,只进行10,000条CCS或者5,000条CCS的测序深度,对于土壤等复杂样品,很难实现稀释曲线饱和,难以检出低丰度物种。
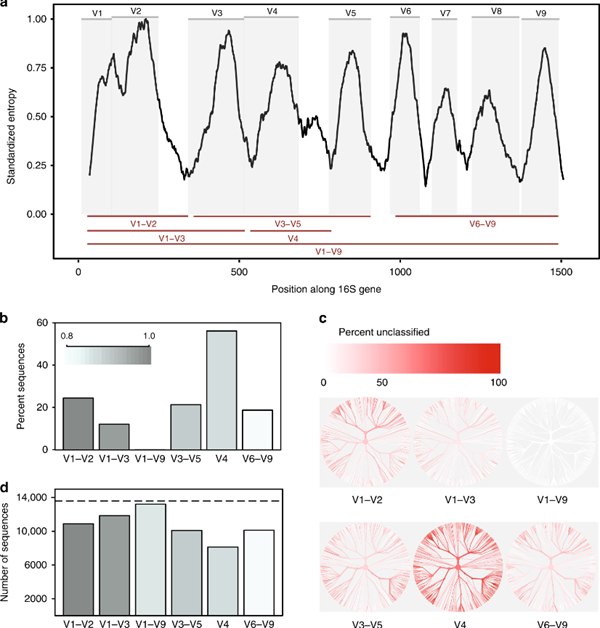
图1 16S可变区比较
高深度三代16S全长是一种基于PacBio Revio测序平台的微生物多样性分析方法,可以对16S rRNA基因进行全长测序,从而实现更高的物种分辨率和准确性。测序数据量高达50,000条CCS甚至100,000条CCS。与传统的二代16S和二代宏基因组测序相比,高深度三代16S全长有以下优势:
相同的测序深度,轻松实现低丰度物种检测。
覆盖16S rRNA基因的所有9个可变区,而二代16S只能测序部分可变区,导致物种注释的不完整和不准确。
可以解决基因组内16S rRNA基因拷贝间的变异问题,而二代16S和二代宏基因组测序无法区分不同拷贝的序列,影响物种和菌株水平的鉴定。
三代16S全长测序技术的应用范畴极为广泛,它不仅在基础科研中发挥着重要作用,更在临床诊断、环境监测、农业以及生物技术开发等多个领域中展示了其独特的价值。
医学研究领域
三代16S全长测序技术已被用于揭示人体微生物组与各类疾病之间的关系。例如,研究表明肠道微生物群落结构的微妙变化与肠道疾病、肥胖、甚至心理健康状况都有着密切联系(Johnsonet al., 2019)。通过全长16S rRNA基因的准确鉴定,研究人员能够更有效地识别出与特定疾病状态相关的微生物群体,进而推动个性化医疗的发展。
图2 肠道微生物与宿主关系(Zhang et al., 2022)
环境科学领域
三代16S全长测序技术在监测水质、土壤微生物多样性以及生物降解过程中的微生物作用方面都有着巨大潜力(Singeret al., 2016)。在大豆根际微生物的研究中发现,PacBio测序的16S全长分辨率更高,而且相对于16S V4区测序,16S全长测序不仅发现了涝害提高了与氮循环相关微生物的相对丰度,而且还发现涝害增加了与磷循环相关微生物的相对丰度(Yu et al., 2022)。
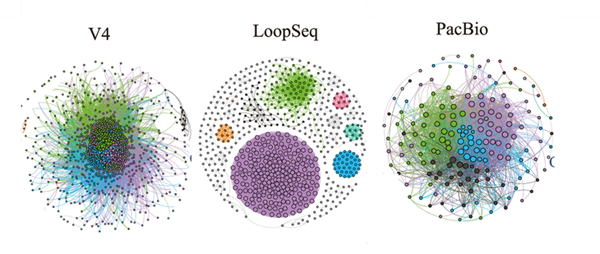
图3 二代16S V4、二代loopseq测序16S全长和PacBio测序16S全长群落内物种相关性网络图(Yu et al., 2022)
农业领域
在农业领域,这项技术可以帮助农业生产者对土壤和植物微生物群落进行深入了解,从而开发出更有效的作物生长促进方法和病害控制策略。例如,分析根际微生物群落结构的变化,可以优化施肥策略,提高作物产量和质量。
生物技术开发
此外,在生物技术开发方面,三代16S全长测序技术的应用也在快速展开。利用这项技术,研究人员可以从自然界中发掘新的微生物资源,包括那些产生新型抗生素、酶或其他生物活性物质的微生物。这不仅有助于生物制药行业的发展,也为环境的生物修复提供了新的策略。
总之,三代16S全长测序技术作为一项革命性的技术,其应用已经并将继续深刻影响着科学研究和实际应用的各个领域。随着未来研究的深入,这项技术有望带来更多令人激动的发现,并进一步扩大其在各个领域中的应用范围。
高深度16S目前有哪些限制?
Silva、greengene、NCBI等数据库大小有限,对于种水平的注释还有待完善;
实验过程需要扩增,如果样品包含酶抑制成分,则会导致扩增失败;
引物有偏好性,且不可避免,目前对于常见细菌的扩增效果比较好,而古菌等特殊原核扩增尚待更多实验验证。
安诺提供什么服务?
16S、ITS、18S全长测序服务;
数据量:平均50,000条CCS/样品,最低40,000条CCS/样品;
质检结果30天质保期;
不定期分析思路分享;
WGCNA、网络图、随机森林预测等个性化分析免费送!
参考文献
Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. Nature. 2012;489(7415): 215-221.
Johnson JS, Spakowicz DJ, Hong BY, et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nature Reviews Microbiology. 2019;17(11): 705-714.
Quince C, Walker AW, Simpson JT, et al. Shotgun metagenomics, from sampling to analysis. Nature Biotechnology. 2017;35(9): 833-844.
Singer E, Wagner M, Woyke T. Capturing the genetic makeup of the active microbiome in situ. Nature Methods. 2016;13(9): 729-731.
Woese CR, Fox GE. Phylogenetic structure of the prokaryotic domain: The primary kingdoms. Nature. 1977;276(5688): 491-494.
Zhang, Chenguang, Liu, Huifeng, Sun, Lei, et al. “ An overview of host-derived molecules that interact with gut microbiota.” iMeta 2023. 2, e88.
Yu T, Cheng L, Liu Q, et al. Effects of Waterlogging on Soybean Rhizosphere Bacterial Community Using V4, LoopSeq, and PacBio 16S rRNA Sequence. Microbiol Spectr. 2022, Feb 23;10(1):e0201121